Venkat Kapil
Ph.D. from EPFL, Switzerland
Oppenheimer Research Fellow @ Department of Chemistry , University of Cambridge, UK
Sydney Harvey Junior Research Fellow @ Churchill College

See projects for more details!
news
| Nov 8, 2019 | Venkat Kapil is awarded the Early Postdoc Mobility Fellowship by the Swiss National Science Foundation. He will spend 18 months in the University of Cambridge as a Visiting Research Fellow and work in close collaboration with Angelos Michaelodes. |
|---|
selected publications
-
 Quantum dynamics using path integral coarse-grainingFélix Musil, Iryna Zaporozhets, Frank Noé, and 2 more authorsAug 2022
Quantum dynamics using path integral coarse-grainingFélix Musil, Iryna Zaporozhets, Frank Noé, and 2 more authorsAug 2022Vibrational spectra of condensed and gas-phase systems containing light nuclei are influenced by their quantum-mechanical behaviour. The quantum dynamics of light nuclei can be approximated by the imaginary time path integral (PI) formulation, but still at a large computational cost that increases sharply with decreasing temperature. By leveraging advances in machine-learned coarse-graining, we develop a PI method with the reduced computational cost of a classical simulation. We also propose a simple temperature elevation scheme to significantly attenuate the artefacts of standard PI approaches and also eliminates the unfavourable temperature scaling of the computational cost.We illustrate the approach, by calculating vibrational spectra using standard models of water molecules and bulk water, demonstrating significant computational savings and dramatically improved accuracy compared to more expensive reference approaches. We believe that our simple, efficient and accurate method could enable routine calculations of vibrational spectra including nuclear quantum effects for a wide range of molecular systems.
@misc{musil_quantum_2022, title = {Quantum dynamics using path integral coarse-graining}, doi = {10.48550/arXiv.2208.06205}, urldate = {2022-08-19}, publisher = {arXiv}, author = {Musil, Félix and Zaporozhets, Iryna and Noé, Frank and Clementi, Cecilia and Kapil, Venkat}, month = aug, year = {2022}, note = {arXiv:2208.06205 [cond-mat, physics:physics, physics:quant-ph]}, keywords = {Condensed Matter - Materials Science, Condensed Matter - Soft Condensed Matter, Condensed Matter - Statistical Mechanics, Physics - Chemical Physics, Quantum Physics}, pi = {true}, mlps = {true} } -
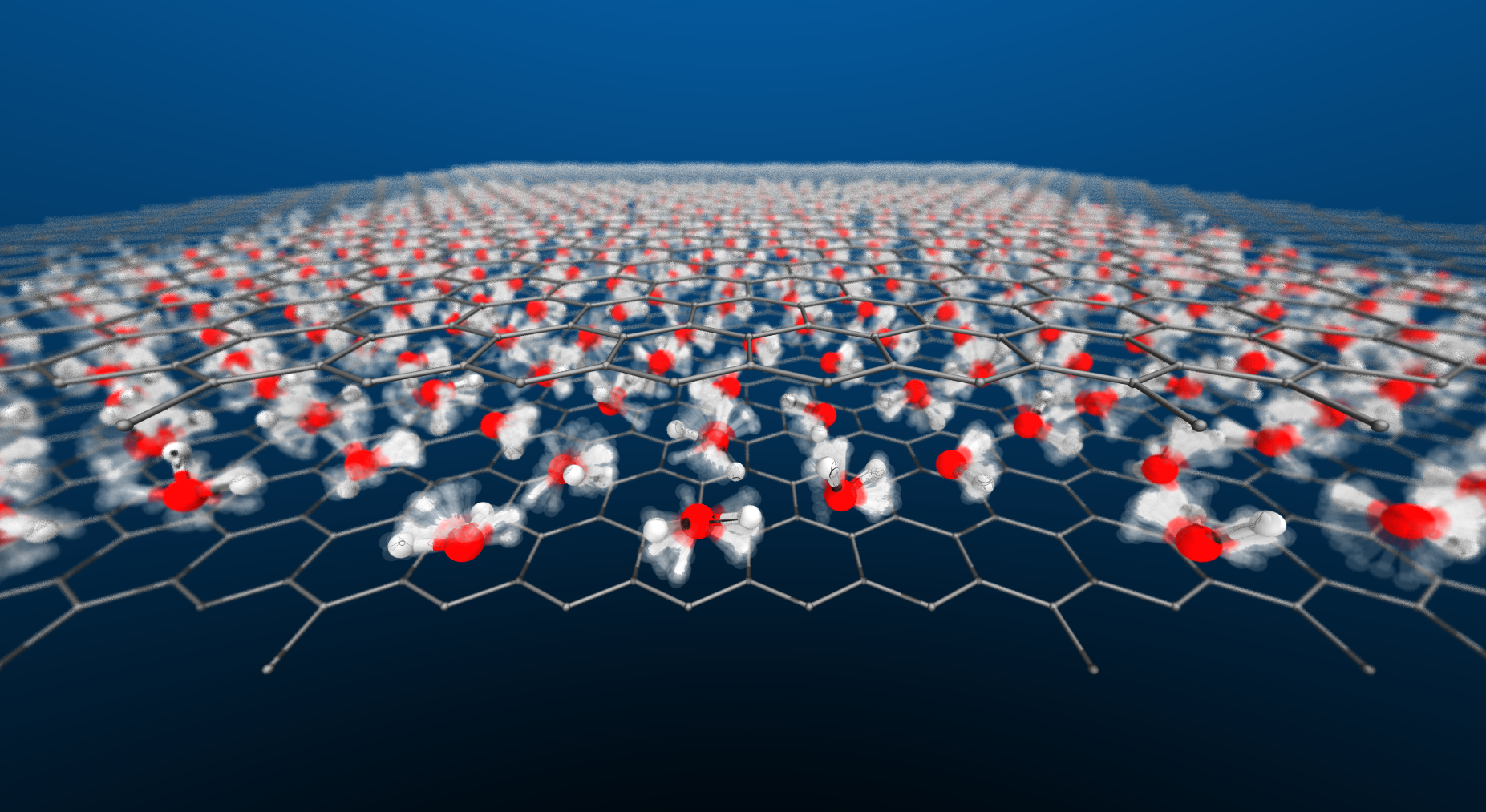 The first-principles phase diagram of monolayer nanoconfined waterVenkat Kapil, Christoph Schran, Andrea Zen, and 3 more authorsJul 2022
The first-principles phase diagram of monolayer nanoconfined waterVenkat Kapil, Christoph Schran, Andrea Zen, and 3 more authorsJul 2022Water in nanoscale cavities is ubiquitous and of central importance to everyday phenomena in geology and biology. However, the properties of nanoscale water can be remarkably different from bulk, as shown e.g., by the anomalously low dielectric constant of water in nanochannels [1], near frictionless water flow [2], or the possible existence of a square ice phase [3]. Such properties suggest that nanoconfined water could be engineered for technological applications in nanouidics [4], electrolyte materials [5], and water desalination [6]. Unfortunately, challenges in experimentally characterising water on the nanoscale and the high cost of first-principles simulations have prevented the molecular level understanding required to control the behavior of water. Here we combine a range of computational approaches to enable a first-principles level investigation of a single layer of water within a graphene-like channel. We find that monolayer water exhibits surprisingly rich and diverse phase behavior that is highly sensitive to temperature and the van der Waals pressure acting within the nanochannel. In addition to multiple molecular phases with melting temperatures varying non-monotonically by over 400 degrees with pressure, we predict a hexatic phase, which is an intermediate between a solid and a liquid, and a superionic phase with a high electrical conductivity exceeding that of battery materials. Notably, this suggests that nanoconfinement could be a promising route towards superionic behavior at easily accessible conditions.
@misc{kapil_first-principles_2022, title = {The first-principles phase diagram of monolayer nanoconfined water}, doi = {10.48550/arXiv.2110.14569}, urldate = {2022-08-19}, publisher = {arXiv}, author = {Kapil, Venkat and Schran, Christoph and Zen, Andrea and Chen, Ji and Pickard, Chris J. and Michaelides, Angelos}, month = jul, year = {2022}, note = {arXiv:2110.14569 [cond-mat]}, keywords = {Condensed Matter - Materials Science, Condensed Matter - Mesoscale and Nanoscale Physics, Condensed Matter - Statistical Mechanics}, ch2o = {true}, mlps = {true}, csp = {true} } -
 i-PI 2.0: A universal force engine for advanced molecular simulationsVenkat Kapil, Mariana Rossi, Ondrej Marsalek, and 24 more authorsComputer Physics Communications Mar 2019
i-PI 2.0: A universal force engine for advanced molecular simulationsVenkat Kapil, Mariana Rossi, Ondrej Marsalek, and 24 more authorsComputer Physics Communications Mar 2019Progress in the atomic-scale modeling of matter over the past decade has been tremendous. This progress has been brought about by improvements in methods for evaluating interatomic forces that work by either solving the electronic structure problem explicitly, or by computing accurate approximations of the solution and by the development of techniques that use the Born–Oppenheimer (BO) forces to move the atoms on the BO potential energy surface. As a consequence of these developments it is now possible to identify stable or metastable states, to sample configurations consistent with the appropriate thermodynamic ensemble, and to estimate the kinetics of reactions and phase transitions. All too often, however, progress is slowed down by the bottleneck associated with implementing new optimization algorithms and/or sampling techniques into the many existing electronic-structure and empirical-potential codes. To address this problem, we are thus releasing a new version of the i-PI software. This piece of software is an easily extensible framework for implementing advanced atomistic simulation techniques using interatomic potentials and forces calculated by an external driver code. While the original version of the code (Ceriotti et al., 2014) was developed with a focus on path integral molecular dynamics techniques, this second release of i-PI not only includes several new advanced path integral methods, but also offers other classes of algorithms. In other words, i-PI is moving towards becoming a universal force engine that is both modular and tightly coupled to the driver codes that evaluate the potential energy surface and its derivatives. Program summary Program Title: i-PI Program Files doi: http://dx.doi.org/10.17632/x792grbm9g.1 Licensing provisions: GPLv3, MIT Programming language: Python External routines/libraries: NumPy Nature of problem: Lowering the implementation barrier to bring state-of-the-art sampling and atomistic modeling techniques to ab initio and empirical potentials programs. Solution method: Advanced sampling methods, including path-integral molecular dynamics techniques, are implemented in a Python interface. Any electronic structure code can be patched to receive the atomic coordinates from the Python interface, and to return the forces and energy that are used to integrate the equations of motion, optimize atomic geometries, etc. Restrictions: This code does not compute interatomic potentials, although the distribution includes sample driver codes that can be used to test different techniques using a few simple model force fields.
@article{kapil_i-pi_2019, title = {i-{PI} 2.0: {A} universal force engine for advanced molecular simulations}, volume = {236}, issn = {0010-4655}, shorttitle = {i-{PI} 2.0}, url = {http://www.sciencedirect.com/science/article/pii/S0010465518303436}, doi = {10.1016/j.cpc.2018.09.020}, language = {en}, urldate = {2019-12-04}, journal = {Computer Physics Communications}, author = {Kapil, Venkat and Rossi, Mariana and Marsalek, Ondrej and Petraglia, Riccardo and Litman, Yair and Spura, Thomas and Cheng, Bingqing and Cuzzocrea, Alice and Meißner, Robert H. and Wilkins, David M. and Helfrecht, Benjamin A. and Juda, Przemysław and Bienvenue, Sébastien P. and Fang, Wei and Kessler, Jan and Poltavsky, Igor and Vandenbrande, Steven and Wieme, Jelle and Corminboeuf, Clemence and Kühne, Thomas D. and Manolopoulos, David E. and Markland, Thomas E. and Richardson, Jeremy O. and Tkatchenko, Alexandre and Tribello, Gareth A. and Van Speybroeck, Veronique and Ceriotti, Michele}, month = mar, year = {2019}, keywords = {Accelerated sampling, Geometry optimizers, Molecular dynamics, Path integral}, pages = {214--223}, pi = {true} } -
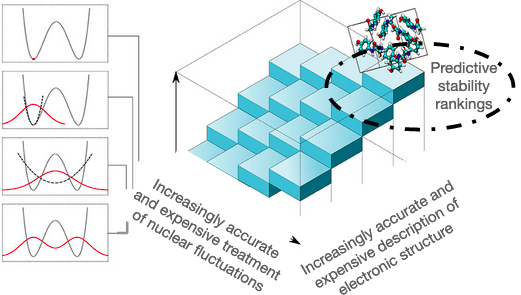 A complete description of thermodynamic stabilities of molecular crystalsVenkat Kapil, and Edgar A. EngelProceedings of the National Academy of Sciences Feb 2022
A complete description of thermodynamic stabilities of molecular crystalsVenkat Kapil, and Edgar A. EngelProceedings of the National Academy of Sciences Feb 2022Predictions of relative stabilities of (competing) molecular crystals are of great technological relevance, most notably for the pharmaceutical industry. However, they present a long-standing challenge for modeling, as often minuscule free energy differences are sensitively affected by the description of electronic structure, the statistical mechanics of the nuclei and the cell, and thermal expansion. The importance of these effects has been individually established, but rigorous free energy calculations for general molecular compounds, which simultaneously account for all effects, have hitherto not been computationally viable. Here we present an efficient “end to end” framework that seamlessly combines state-of-the art electronic structure calculations, machine-learning potentials, and advanced free energy methods to calculate ab initio Gibbs free energies for general organic molecular materials. The facile generation of machine-learning potentials for a diverse set of polymorphic compounds—benzene, glycine, and succinic acid—and predictions of thermodynamic stabilities in qualitative and quantitative agreement with experiments highlight that predictive thermodynamic studies of industrially relevant molecular materials are no longer a daunting task.
@article{kapil_complete_2022, title = {A complete description of thermodynamic stabilities of molecular crystals}, volume = {119}, copyright = {Copyright © 2022 the Author(s). Published by PNAS.. https://creativecommons.org/licenses/by/4.0/This open access article is distributed under Creative Commons Attribution License 4.0 (CC BY).}, issn = {0027-8424, 1091-6490}, url = {https://www.pnas.org/content/119/6/e2111769119}, doi = {10.1073/pnas.2111769119}, language = {en}, number = {6}, urldate = {2022-02-10}, journal = {Proceedings of the National Academy of Sciences}, author = {Kapil, Venkat and Engel, Edgar A.}, month = feb, year = {2022}, pmid = {35131847}, keywords = {Condensed Matter - Materials Science, Condensed Matter - Statistical Mechanics, ab initio thermodynamics, machine learning, polymorphism, statistical mechanics}, pi = {true}, mlps = {true}, csp = {true} } -
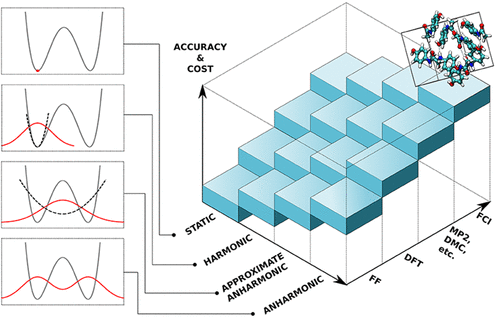 Assessment of Approximate Methods for Anharmonic Free EnergiesVenkat Kapil, Edgar Engel, Mariana Rossi, and 1 more authorJournal of Chemical Theory and Computation Nov 2019
Assessment of Approximate Methods for Anharmonic Free EnergiesVenkat Kapil, Edgar Engel, Mariana Rossi, and 1 more authorJournal of Chemical Theory and Computation Nov 2019Quantitative evaluation of the thermodynamic properties of materials—most notably their stability, as measured by the free energy—must take into account the role of thermal and zero-point energy fluctuations. While these effects can easily be estimated within a harmonic approximation, corrections arising from the anharmonic nature of the interatomic potential are often crucial and require computationally costly path integral simulations to obtain results that are essentially exact for a given potential. Consequently, different approximate frameworks for computing affordable estimates of the anharmonic free energies have been developed over the years. Understanding which of the approximations involved are justified for a given system, and therefore choosing the most suitable method, is complicated by the lack of comparative benchmarks. To facilitate this choice we assess the accuracy and efficiency of some of the most commonly used approximate methods: the independent mode framework, the vibrational self-consistent field, and self-consistent phonons. We compare the anharmonic correction to the Helmholtz free energy against reference path integral calculations. These benchmarks are performed for a diverse set of systems, ranging from simple weakly anharmonic solids to flexible molecular crystals with freely rotating units. The results suggest that, for simple solids such as allotropes of carbon, these methods yield results that are in excellent agreement with the reference calculations, at a considerably lower computational cost. For more complex molecular systems such as polymorphs of ice and paracetamol the methods do not consistently provide a reliable approximation of the anharmonic correction. Despite substantial cancellation of errors when comparing the stability of different phases, we do not observe a systematic improvement over the harmonic approximation even for relative free energies. We conclude that, at least for the classes of materials considered here, efforts toward obtaining computationally feasible anharmonic free energies should therefore be directed toward reducing the expense of path integral methods.
@article{kapil_assessment_2019, title = {Assessment of {Approximate} {Methods} for {Anharmonic} {Free} {Energies}}, volume = {15}, issn = {1549-9618}, url = {https://doi.org/10.1021/acs.jctc.9b00596}, doi = {10.1021/acs.jctc.9b00596}, number = {11}, urldate = {2020-01-22}, journal = {Journal of Chemical Theory and Computation}, author = {Kapil, Venkat and Engel, Edgar and Rossi, Mariana and Ceriotti, Michele}, month = nov, year = {2019}, pages = {5845--5857}, csp = {true} } -
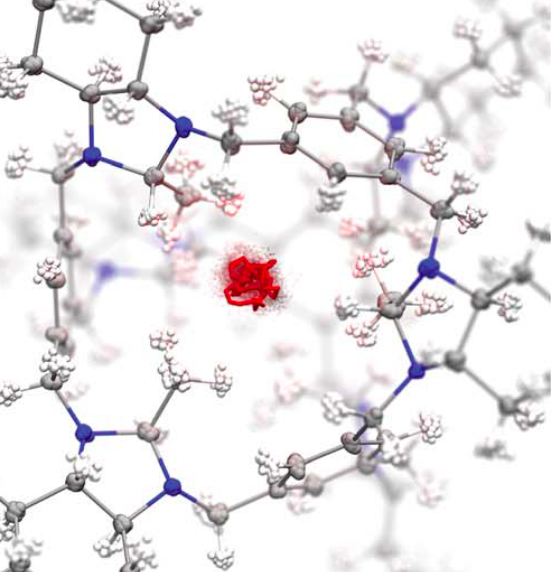 Barely porous organic cages for hydrogen isotope separationMing Liu, Linda Zhang, Marc A. Little, and 13 more authorsScience Nov 2019
Barely porous organic cages for hydrogen isotope separationMing Liu, Linda Zhang, Marc A. Little, and 13 more authorsScience Nov 2019Quantum sieves for hydrogen isotopes One method for improving the efficiency of separation of hydrogen from deuterium (D) is to exploit kinetic quantum sieving with nanoporous solids. This method requires ultrafine pore apertures (around 3 angstroms), which usually leads to low pore volumes and low D2 adsorption capacities. Liu et al. used organic synthesis to tune the pore size of the internal cavities of organic cage molecules. A hybrid cocrystal contained both a small-pore cage that imparted high selectivity and a larger-pore cage that enabled high D2 uptake. Science, this issue p. 613 The separation of hydrogen isotopes for applications such as nuclear fusion is a major challenge. Current technologies are energy intensive and inefficient. Nanoporous materials have the potential to separate hydrogen isotopes by kinetic quantum sieving, but high separation selectivity tends to correlate with low adsorption capacity, which can prohibit process scale-up. In this study, we use organic synthesis to modify the internal cavities of cage molecules to produce hybrid materials that are excellent quantum sieves. By combining small-pore and large-pore cages together in a single solid, we produce a material with optimal separation performance that combines an excellent deuterium/hydrogen selectivity (8.0) with a high deuterium uptake (4.7 millimoles per gram). Cocrystals of modified molecular organic crystals can separate hydrogen from deuterium through kinetic quantum sieving. Cocrystals of modified molecular organic crystals can separate hydrogen from deuterium through kinetic quantum sieving.
@article{liu_barely_2019, title = {Barely porous organic cages for hydrogen isotope separation}, volume = {366}, copyright = {Copyright © 2019 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works. http://www.sciencemag.org/about/science-licenses-journal-article-reuseThis is an article distributed under the terms of the Science Journals Default License.}, issn = {0036-8075, 1095-9203}, url = {https://science.sciencemag.org/content/366/6465/613}, doi = {10.1126/science.aax7427}, language = {en}, number = {6465}, urldate = {2019-11-18}, journal = {Science}, author = {Liu, Ming and Zhang, Linda and Little, Marc A. and Kapil, Venkat and Ceriotti, Michele and Yang, Siyuan and Ding, Lifeng and Holden, Daniel L. and Balderas-Xicohténcatl, Rafael and He, Donglin and Clowes, Rob and Chong, Samantha Y. and Schütz, Gisela and Chen, Linjiang and Hirscher, Michael and Cooper, Andrew I.}, month = nov, year = {2019}, pmid = {31672893}, pages = {613--620}, pi = {true} }