crystal structure prediction
We develop approaches to predict the first-principles thermodynamics stability (i.e. its “absolute” free energy) by combining quantum-mechanics, machine-learning and high-level electronic structure methods.
Selected Publications
-
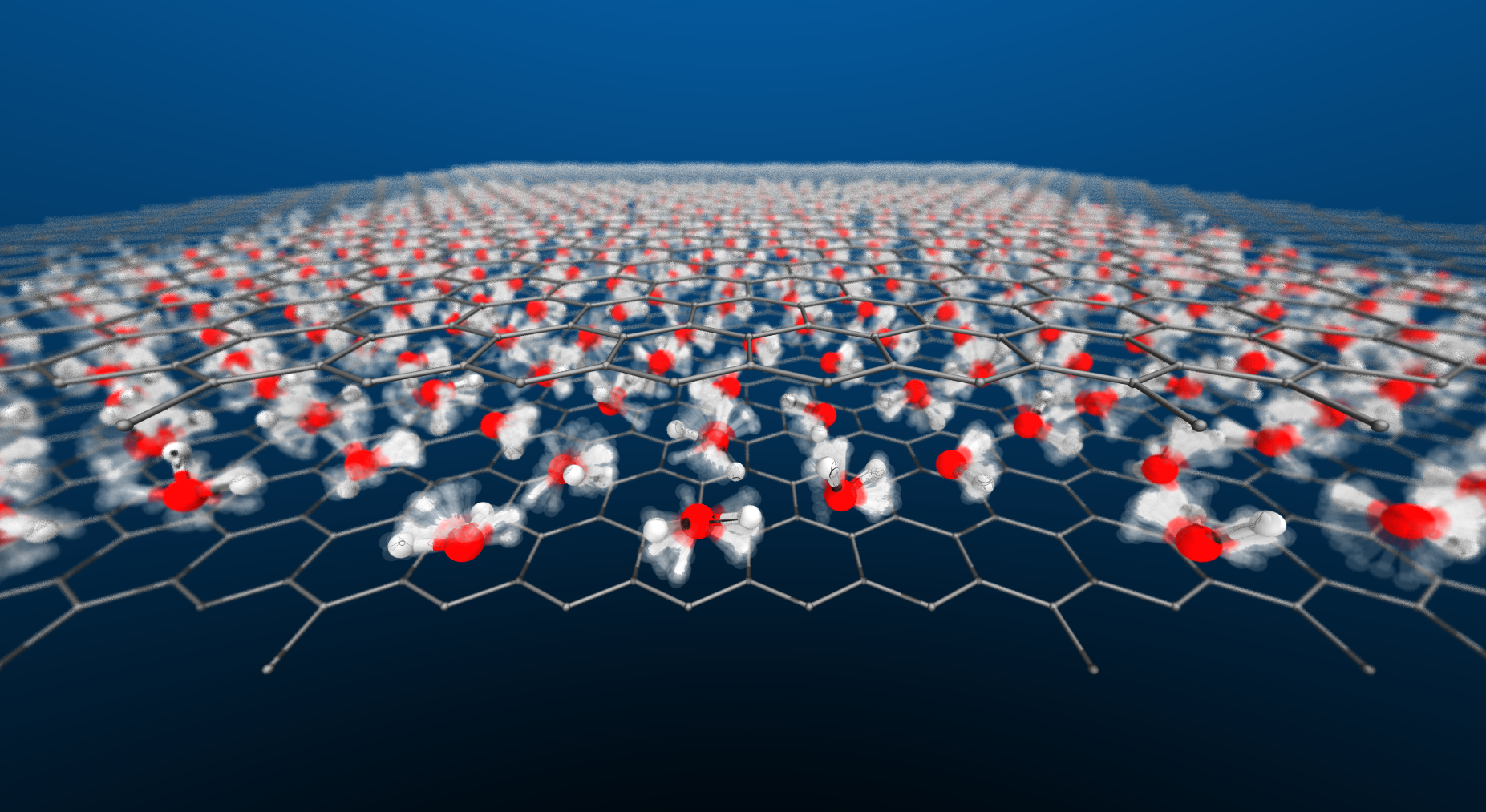 The first-principles phase diagram of monolayer nanoconfined waterVenkat Kapil, Christoph Schran, Andrea Zen, and 3 more authorsJul 2022
The first-principles phase diagram of monolayer nanoconfined waterVenkat Kapil, Christoph Schran, Andrea Zen, and 3 more authorsJul 2022Water in nanoscale cavities is ubiquitous and of central importance to everyday phenomena in geology and biology. However, the properties of nanoscale water can be remarkably different from bulk, as shown e.g., by the anomalously low dielectric constant of water in nanochannels [1], near frictionless water flow [2], or the possible existence of a square ice phase [3]. Such properties suggest that nanoconfined water could be engineered for technological applications in nanouidics [4], electrolyte materials [5], and water desalination [6]. Unfortunately, challenges in experimentally characterising water on the nanoscale and the high cost of first-principles simulations have prevented the molecular level understanding required to control the behavior of water. Here we combine a range of computational approaches to enable a first-principles level investigation of a single layer of water within a graphene-like channel. We find that monolayer water exhibits surprisingly rich and diverse phase behavior that is highly sensitive to temperature and the van der Waals pressure acting within the nanochannel. In addition to multiple molecular phases with melting temperatures varying non-monotonically by over 400 degrees with pressure, we predict a hexatic phase, which is an intermediate between a solid and a liquid, and a superionic phase with a high electrical conductivity exceeding that of battery materials. Notably, this suggests that nanoconfinement could be a promising route towards superionic behavior at easily accessible conditions.
@misc{kapil_first-principles_2022, title = {The first-principles phase diagram of monolayer nanoconfined water}, doi = {10.48550/arXiv.2110.14569}, urldate = {2022-08-19}, publisher = {arXiv}, author = {Kapil, Venkat and Schran, Christoph and Zen, Andrea and Chen, Ji and Pickard, Chris J. and Michaelides, Angelos}, month = jul, year = {2022}, note = {arXiv:2110.14569 [cond-mat]}, keywords = {Condensed Matter - Materials Science, Condensed Matter - Mesoscale and Nanoscale Physics, Condensed Matter - Statistical Mechanics}, ch2o = {true}, mlps = {true}, csp = {true} } -
 General embedded cluster protocol for accurate modeling of oxygen vacancies in metal-oxidesBenjamin X. Shi, Venkat Kapil, Andrea Zen, and 3 more authorsThe Journal of Chemical Physics Mar 2022
General embedded cluster protocol for accurate modeling of oxygen vacancies in metal-oxidesBenjamin X. Shi, Venkat Kapil, Andrea Zen, and 3 more authorsThe Journal of Chemical Physics Mar 2022The O vacancy (Ov) formation energy, EOv, is an important property of a metal-oxide, governing its performance in applications such as fuel cells or heterogeneous catalysis. These defects are routinely studied with density functional theory (DFT). However, it is well-recognized that standard DFT formulations (e.g., the generalized gradient approximation) are insufficient for modeling the Ov, requiring higher levels of theory. The embedded cluster method offers a promising approach to compute EOv accurately, giving access to all electronic structure methods. Central to this approach is the construction of quantum(-mechanically treated) clusters placed within suitable embedding environments. Unfortunately, current approaches to constructing the quantum clusters either require large system sizes, preventing application of high-level methods, or require significant manual input, preventing investigations of multiple systems simultaneously. In this work, we present a systematic and general quantum cluster design protocol that can determine small converged quantum clusters for studying the Ov in metal-oxides with accurate methods, such as local coupled cluster with single, double, and perturbative triple excitations. We apply this protocol to study the Ov in the bulk and surface planes of rutile TiO2 and rock salt MgO, producing the first accurate and well-converged determinations of EOv with this method. These reference values are used to benchmark exchange–correlation functionals in DFT, and we find that all the studied functionals underestimate EOv, with the average error decreasing along the rungs of Jacob’s ladder. This protocol is automatable for high-throughput calculations and can be generalized to study other point defects or adsorbates.
@article{shi_general_2022, title = {General embedded cluster protocol for accurate modeling of oxygen vacancies in metal-oxides}, volume = {156}, issn = {0021-9606}, url = {https://aip.scitation.org/doi/full/10.1063/5.0087031}, doi = {10.1063/5.0087031}, number = {12}, urldate = {2022-08-19}, journal = {The Journal of Chemical Physics}, author = {Shi, Benjamin X. and Kapil, Venkat and Zen, Andrea and Chen, Ji and Alavi, Ali and Michaelides, Angelos}, month = mar, year = {2022}, pages = {124704}, csp = {true} } -
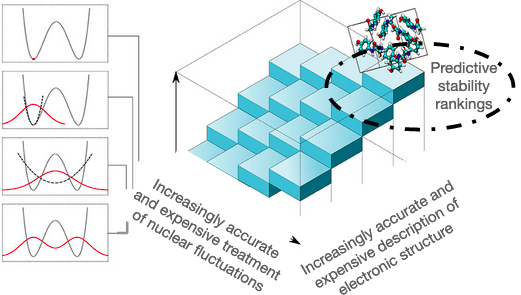 A complete description of thermodynamic stabilities of molecular crystalsVenkat Kapil, and Edgar A. EngelProceedings of the National Academy of Sciences Feb 2022
A complete description of thermodynamic stabilities of molecular crystalsVenkat Kapil, and Edgar A. EngelProceedings of the National Academy of Sciences Feb 2022Predictions of relative stabilities of (competing) molecular crystals are of great technological relevance, most notably for the pharmaceutical industry. However, they present a long-standing challenge for modeling, as often minuscule free energy differences are sensitively affected by the description of electronic structure, the statistical mechanics of the nuclei and the cell, and thermal expansion. The importance of these effects has been individually established, but rigorous free energy calculations for general molecular compounds, which simultaneously account for all effects, have hitherto not been computationally viable. Here we present an efficient “end to end” framework that seamlessly combines state-of-the art electronic structure calculations, machine-learning potentials, and advanced free energy methods to calculate ab initio Gibbs free energies for general organic molecular materials. The facile generation of machine-learning potentials for a diverse set of polymorphic compounds—benzene, glycine, and succinic acid—and predictions of thermodynamic stabilities in qualitative and quantitative agreement with experiments highlight that predictive thermodynamic studies of industrially relevant molecular materials are no longer a daunting task.
@article{kapil_complete_2022, title = {A complete description of thermodynamic stabilities of molecular crystals}, volume = {119}, copyright = {Copyright © 2022 the Author(s). Published by PNAS.. https://creativecommons.org/licenses/by/4.0/This open access article is distributed under Creative Commons Attribution License 4.0 (CC BY).}, issn = {0027-8424, 1091-6490}, url = {https://www.pnas.org/content/119/6/e2111769119}, doi = {10.1073/pnas.2111769119}, language = {en}, number = {6}, urldate = {2022-02-10}, journal = {Proceedings of the National Academy of Sciences}, author = {Kapil, Venkat and Engel, Edgar A.}, month = feb, year = {2022}, pmid = {35131847}, keywords = {Condensed Matter - Materials Science, Condensed Matter - Statistical Mechanics, ab initio thermodynamics, machine learning, polymorphism, statistical mechanics}, pi = {true}, mlps = {true}, csp = {true} } -
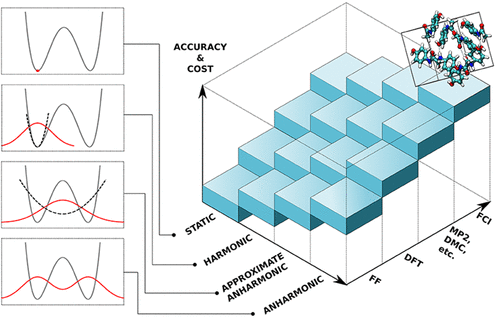 Assessment of Approximate Methods for Anharmonic Free EnergiesVenkat Kapil, Edgar Engel, Mariana Rossi, and 1 more authorJournal of Chemical Theory and Computation Nov 2019
Assessment of Approximate Methods for Anharmonic Free EnergiesVenkat Kapil, Edgar Engel, Mariana Rossi, and 1 more authorJournal of Chemical Theory and Computation Nov 2019Quantitative evaluation of the thermodynamic properties of materials—most notably their stability, as measured by the free energy—must take into account the role of thermal and zero-point energy fluctuations. While these effects can easily be estimated within a harmonic approximation, corrections arising from the anharmonic nature of the interatomic potential are often crucial and require computationally costly path integral simulations to obtain results that are essentially exact for a given potential. Consequently, different approximate frameworks for computing affordable estimates of the anharmonic free energies have been developed over the years. Understanding which of the approximations involved are justified for a given system, and therefore choosing the most suitable method, is complicated by the lack of comparative benchmarks. To facilitate this choice we assess the accuracy and efficiency of some of the most commonly used approximate methods: the independent mode framework, the vibrational self-consistent field, and self-consistent phonons. We compare the anharmonic correction to the Helmholtz free energy against reference path integral calculations. These benchmarks are performed for a diverse set of systems, ranging from simple weakly anharmonic solids to flexible molecular crystals with freely rotating units. The results suggest that, for simple solids such as allotropes of carbon, these methods yield results that are in excellent agreement with the reference calculations, at a considerably lower computational cost. For more complex molecular systems such as polymorphs of ice and paracetamol the methods do not consistently provide a reliable approximation of the anharmonic correction. Despite substantial cancellation of errors when comparing the stability of different phases, we do not observe a systematic improvement over the harmonic approximation even for relative free energies. We conclude that, at least for the classes of materials considered here, efforts toward obtaining computationally feasible anharmonic free energies should therefore be directed toward reducing the expense of path integral methods.
@article{kapil_assessment_2019, title = {Assessment of {Approximate} {Methods} for {Anharmonic} {Free} {Energies}}, volume = {15}, issn = {1549-9618}, url = {https://doi.org/10.1021/acs.jctc.9b00596}, doi = {10.1021/acs.jctc.9b00596}, number = {11}, urldate = {2020-01-22}, journal = {Journal of Chemical Theory and Computation}, author = {Kapil, Venkat and Engel, Edgar and Rossi, Mariana and Ceriotti, Michele}, month = nov, year = {2019}, pages = {5845--5857}, csp = {true} }