machine learning methods
We develop approaches to expidite the production of machine-learning potentials, machine-learning models for electronic properties and associated uncertainty estimates.
Selected Publications
-
 Quantum dynamics using path integral coarse-grainingFélix Musil, Iryna Zaporozhets, Frank Noé, and 2 more authorsAug 2022
Quantum dynamics using path integral coarse-grainingFélix Musil, Iryna Zaporozhets, Frank Noé, and 2 more authorsAug 2022Vibrational spectra of condensed and gas-phase systems containing light nuclei are influenced by their quantum-mechanical behaviour. The quantum dynamics of light nuclei can be approximated by the imaginary time path integral (PI) formulation, but still at a large computational cost that increases sharply with decreasing temperature. By leveraging advances in machine-learned coarse-graining, we develop a PI method with the reduced computational cost of a classical simulation. We also propose a simple temperature elevation scheme to significantly attenuate the artefacts of standard PI approaches and also eliminates the unfavourable temperature scaling of the computational cost.We illustrate the approach, by calculating vibrational spectra using standard models of water molecules and bulk water, demonstrating significant computational savings and dramatically improved accuracy compared to more expensive reference approaches. We believe that our simple, efficient and accurate method could enable routine calculations of vibrational spectra including nuclear quantum effects for a wide range of molecular systems.
@misc{musil_quantum_2022, title = {Quantum dynamics using path integral coarse-graining}, doi = {10.48550/arXiv.2208.06205}, urldate = {2022-08-19}, publisher = {arXiv}, author = {Musil, Félix and Zaporozhets, Iryna and Noé, Frank and Clementi, Cecilia and Kapil, Venkat}, month = aug, year = {2022}, note = {arXiv:2208.06205 [cond-mat, physics:physics, physics:quant-ph]}, keywords = {Condensed Matter - Materials Science, Condensed Matter - Soft Condensed Matter, Condensed Matter - Statistical Mechanics, Physics - Chemical Physics, Quantum Physics}, pi = {true}, mlps = {true} } -
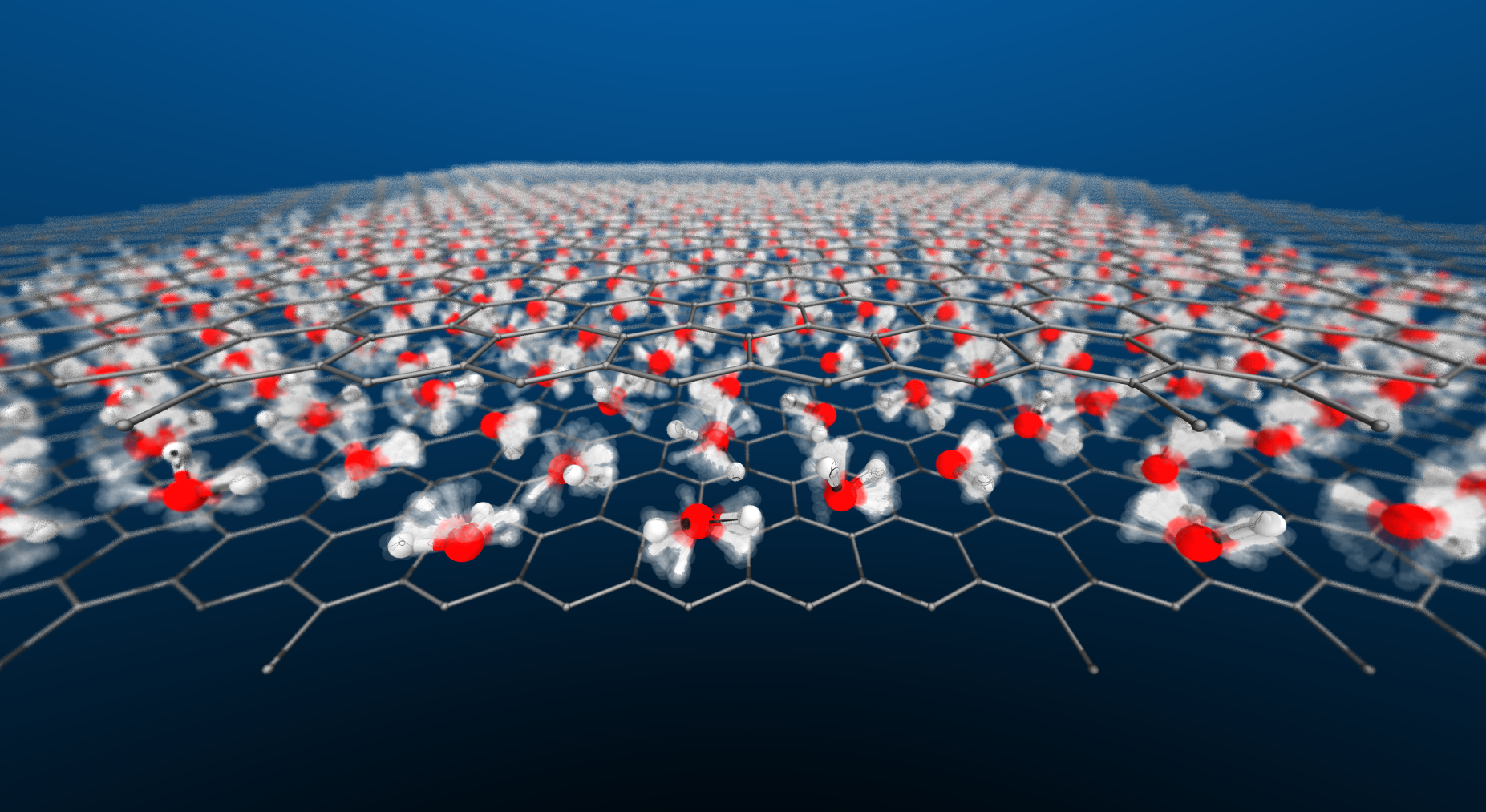 The first-principles phase diagram of monolayer nanoconfined waterVenkat Kapil, Christoph Schran, Andrea Zen, and 3 more authorsJul 2022
The first-principles phase diagram of monolayer nanoconfined waterVenkat Kapil, Christoph Schran, Andrea Zen, and 3 more authorsJul 2022Water in nanoscale cavities is ubiquitous and of central importance to everyday phenomena in geology and biology. However, the properties of nanoscale water can be remarkably different from bulk, as shown e.g., by the anomalously low dielectric constant of water in nanochannels [1], near frictionless water flow [2], or the possible existence of a square ice phase [3]. Such properties suggest that nanoconfined water could be engineered for technological applications in nanouidics [4], electrolyte materials [5], and water desalination [6]. Unfortunately, challenges in experimentally characterising water on the nanoscale and the high cost of first-principles simulations have prevented the molecular level understanding required to control the behavior of water. Here we combine a range of computational approaches to enable a first-principles level investigation of a single layer of water within a graphene-like channel. We find that monolayer water exhibits surprisingly rich and diverse phase behavior that is highly sensitive to temperature and the van der Waals pressure acting within the nanochannel. In addition to multiple molecular phases with melting temperatures varying non-monotonically by over 400 degrees with pressure, we predict a hexatic phase, which is an intermediate between a solid and a liquid, and a superionic phase with a high electrical conductivity exceeding that of battery materials. Notably, this suggests that nanoconfinement could be a promising route towards superionic behavior at easily accessible conditions.
@misc{kapil_first-principles_2022, title = {The first-principles phase diagram of monolayer nanoconfined water}, doi = {10.48550/arXiv.2110.14569}, urldate = {2022-08-19}, publisher = {arXiv}, author = {Kapil, Venkat and Schran, Christoph and Zen, Andrea and Chen, Ji and Pickard, Chris J. and Michaelides, Angelos}, month = jul, year = {2022}, note = {arXiv:2110.14569 [cond-mat]}, keywords = {Condensed Matter - Materials Science, Condensed Matter - Mesoscale and Nanoscale Physics, Condensed Matter - Statistical Mechanics}, ch2o = {true}, mlps = {true}, csp = {true} } -
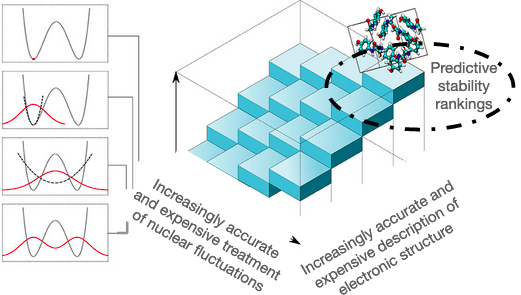 A complete description of thermodynamic stabilities of molecular crystalsVenkat Kapil, and Edgar A. EngelProceedings of the National Academy of Sciences Feb 2022
A complete description of thermodynamic stabilities of molecular crystalsVenkat Kapil, and Edgar A. EngelProceedings of the National Academy of Sciences Feb 2022Predictions of relative stabilities of (competing) molecular crystals are of great technological relevance, most notably for the pharmaceutical industry. However, they present a long-standing challenge for modeling, as often minuscule free energy differences are sensitively affected by the description of electronic structure, the statistical mechanics of the nuclei and the cell, and thermal expansion. The importance of these effects has been individually established, but rigorous free energy calculations for general molecular compounds, which simultaneously account for all effects, have hitherto not been computationally viable. Here we present an efficient “end to end” framework that seamlessly combines state-of-the art electronic structure calculations, machine-learning potentials, and advanced free energy methods to calculate ab initio Gibbs free energies for general organic molecular materials. The facile generation of machine-learning potentials for a diverse set of polymorphic compounds—benzene, glycine, and succinic acid—and predictions of thermodynamic stabilities in qualitative and quantitative agreement with experiments highlight that predictive thermodynamic studies of industrially relevant molecular materials are no longer a daunting task.
@article{kapil_complete_2022, title = {A complete description of thermodynamic stabilities of molecular crystals}, volume = {119}, copyright = {Copyright © 2022 the Author(s). Published by PNAS.. https://creativecommons.org/licenses/by/4.0/This open access article is distributed under Creative Commons Attribution License 4.0 (CC BY).}, issn = {0027-8424, 1091-6490}, url = {https://www.pnas.org/content/119/6/e2111769119}, doi = {10.1073/pnas.2111769119}, language = {en}, number = {6}, urldate = {2022-02-10}, journal = {Proceedings of the National Academy of Sciences}, author = {Kapil, Venkat and Engel, Edgar A.}, month = feb, year = {2022}, pmid = {35131847}, keywords = {Condensed Matter - Materials Science, Condensed Matter - Statistical Mechanics, ab initio thermodynamics, machine learning, polymorphism, statistical mechanics}, pi = {true}, mlps = {true}, csp = {true} } -
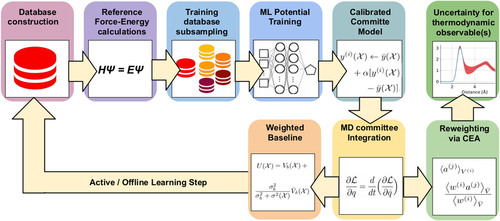 Uncertainty estimation for molecular dynamics and samplingGiulio Imbalzano, Yongbin Zhuang, Venkat Kapil, and 4 more authorsThe Journal of Chemical Physics Feb 2021
Uncertainty estimation for molecular dynamics and samplingGiulio Imbalzano, Yongbin Zhuang, Venkat Kapil, and 4 more authorsThe Journal of Chemical Physics Feb 2021Machine-learning models have emerged as a very effective strategy to sidestep time-consuming electronic-structure calculations, enabling accurate simulations of greater size, time scale, and complexity. Given the interpolative nature of these models, the reliability of predictions depends on the position in phase space, and it is crucial to obtain an estimate of the error that derives from the finite number of reference structures included during model training. When using a machine-learning potential to sample a finite-temperature ensemble, the uncertainty on individual configurations translates into an error on thermodynamic averages and leads to a loss of accuracy when the simulation enters a previously unexplored region. Here, we discuss how uncertainty quantification can be used, together with a baseline energy model, or a more robust but less accurate interatomic potential, to obtain more resilient simulations and to support active-learning strategies. Furthermore, we introduce an on-the-fly reweighing scheme that makes it possible to estimate the uncertainty in thermodynamic averages extracted from long trajectories. We present examples covering different types of structural and thermodynamic properties and systems as diverse as water and liquid gallium.
@article{imbalzano_uncertainty_2021, title = {Uncertainty estimation for molecular dynamics and sampling}, volume = {154}, issn = {0021-9606}, url = {https://aip.scitation.org/doi/10.1063/5.0036522}, doi = {10.1063/5.0036522}, number = {7}, urldate = {2021-05-31}, journal = {The Journal of Chemical Physics}, author = {Imbalzano, Giulio and Zhuang, Yongbin and Kapil, Venkat and Rossi, Kevin and Engel, Edgar A. and Grasselli, Federico and Ceriotti, Michele}, month = feb, year = {2021}, pages = {074102}, mlps = {true} }