publications
publications by categories in reversed chronological order. generated by jekyll-scholar.
2022
-
 Quantum dynamics using path integral coarse-grainingFélix Musil, Iryna Zaporozhets, Frank Noé, and 2 more authorsAug 2022
Quantum dynamics using path integral coarse-grainingFélix Musil, Iryna Zaporozhets, Frank Noé, and 2 more authorsAug 2022Vibrational spectra of condensed and gas-phase systems containing light nuclei are influenced by their quantum-mechanical behaviour. The quantum dynamics of light nuclei can be approximated by the imaginary time path integral (PI) formulation, but still at a large computational cost that increases sharply with decreasing temperature. By leveraging advances in machine-learned coarse-graining, we develop a PI method with the reduced computational cost of a classical simulation. We also propose a simple temperature elevation scheme to significantly attenuate the artefacts of standard PI approaches and also eliminates the unfavourable temperature scaling of the computational cost.We illustrate the approach, by calculating vibrational spectra using standard models of water molecules and bulk water, demonstrating significant computational savings and dramatically improved accuracy compared to more expensive reference approaches. We believe that our simple, efficient and accurate method could enable routine calculations of vibrational spectra including nuclear quantum effects for a wide range of molecular systems.
@misc{musil_quantum_2022, title = {Quantum dynamics using path integral coarse-graining}, doi = {10.48550/arXiv.2208.06205}, urldate = {2022-08-19}, publisher = {arXiv}, author = {Musil, Félix and Zaporozhets, Iryna and Noé, Frank and Clementi, Cecilia and Kapil, Venkat}, month = aug, year = {2022}, note = {arXiv:2208.06205 [cond-mat, physics:physics, physics:quant-ph]}, keywords = {Condensed Matter - Materials Science, Condensed Matter - Soft Condensed Matter, Condensed Matter - Statistical Mechanics, Physics - Chemical Physics, Quantum Physics}, pi = {true}, mlps = {true} } -
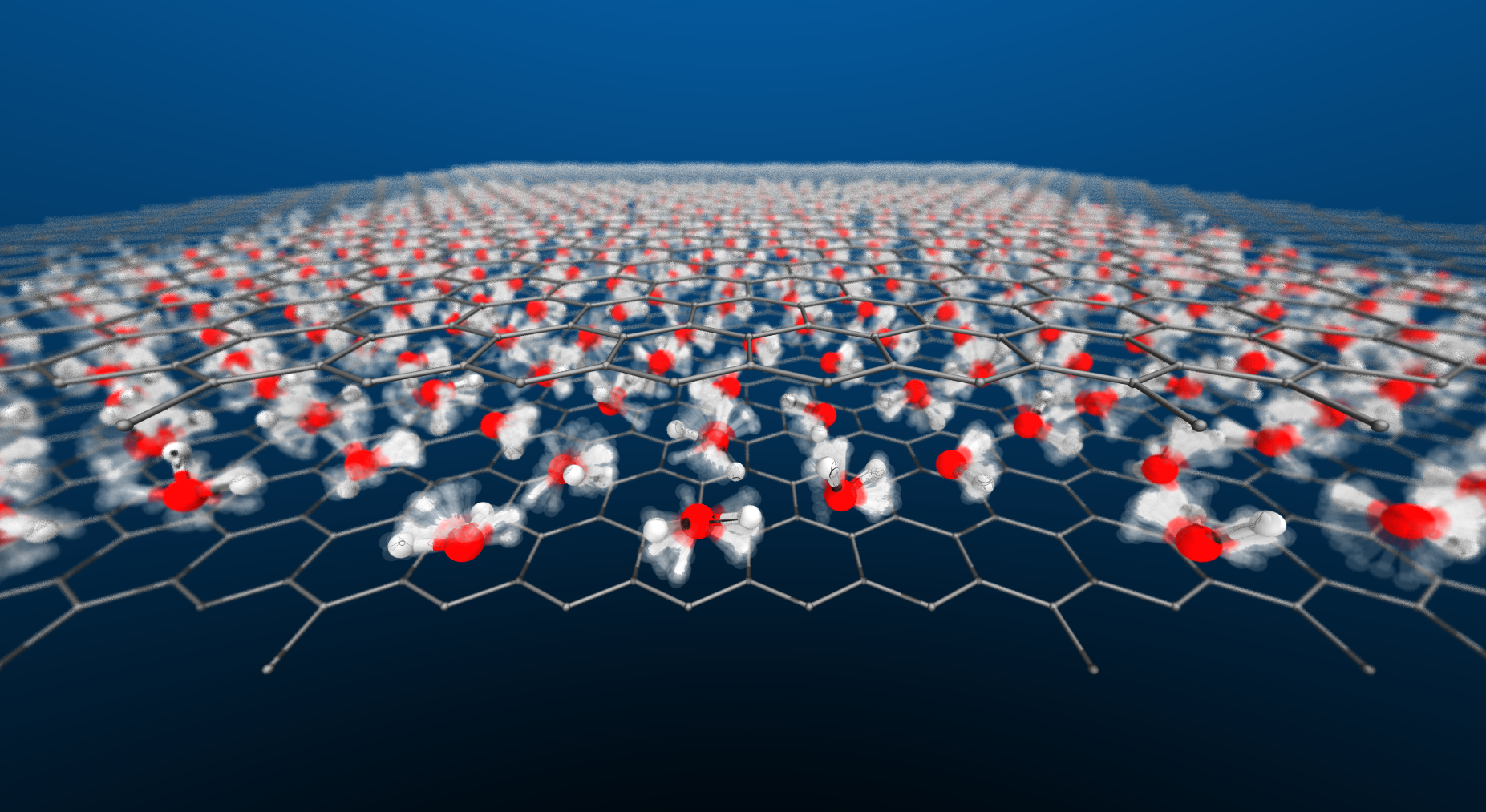 The first-principles phase diagram of monolayer nanoconfined waterVenkat Kapil, Christoph Schran, Andrea Zen, and 3 more authorsJul 2022
The first-principles phase diagram of monolayer nanoconfined waterVenkat Kapil, Christoph Schran, Andrea Zen, and 3 more authorsJul 2022Water in nanoscale cavities is ubiquitous and of central importance to everyday phenomena in geology and biology. However, the properties of nanoscale water can be remarkably different from bulk, as shown e.g., by the anomalously low dielectric constant of water in nanochannels [1], near frictionless water flow [2], or the possible existence of a square ice phase [3]. Such properties suggest that nanoconfined water could be engineered for technological applications in nanouidics [4], electrolyte materials [5], and water desalination [6]. Unfortunately, challenges in experimentally characterising water on the nanoscale and the high cost of first-principles simulations have prevented the molecular level understanding required to control the behavior of water. Here we combine a range of computational approaches to enable a first-principles level investigation of a single layer of water within a graphene-like channel. We find that monolayer water exhibits surprisingly rich and diverse phase behavior that is highly sensitive to temperature and the van der Waals pressure acting within the nanochannel. In addition to multiple molecular phases with melting temperatures varying non-monotonically by over 400 degrees with pressure, we predict a hexatic phase, which is an intermediate between a solid and a liquid, and a superionic phase with a high electrical conductivity exceeding that of battery materials. Notably, this suggests that nanoconfinement could be a promising route towards superionic behavior at easily accessible conditions.
@misc{kapil_first-principles_2022, title = {The first-principles phase diagram of monolayer nanoconfined water}, doi = {10.48550/arXiv.2110.14569}, urldate = {2022-08-19}, publisher = {arXiv}, author = {Kapil, Venkat and Schran, Christoph and Zen, Andrea and Chen, Ji and Pickard, Chris J. and Michaelides, Angelos}, month = jul, year = {2022}, note = {arXiv:2110.14569 [cond-mat]}, keywords = {Condensed Matter - Materials Science, Condensed Matter - Mesoscale and Nanoscale Physics, Condensed Matter - Statistical Mechanics}, ch2o = {true}, mlps = {true}, csp = {true} } -
 General embedded cluster protocol for accurate modeling of oxygen vacancies in metal-oxidesBenjamin X. Shi, Venkat Kapil, Andrea Zen, and 3 more authorsThe Journal of Chemical Physics Mar 2022
General embedded cluster protocol for accurate modeling of oxygen vacancies in metal-oxidesBenjamin X. Shi, Venkat Kapil, Andrea Zen, and 3 more authorsThe Journal of Chemical Physics Mar 2022The O vacancy (Ov) formation energy, EOv, is an important property of a metal-oxide, governing its performance in applications such as fuel cells or heterogeneous catalysis. These defects are routinely studied with density functional theory (DFT). However, it is well-recognized that standard DFT formulations (e.g., the generalized gradient approximation) are insufficient for modeling the Ov, requiring higher levels of theory. The embedded cluster method offers a promising approach to compute EOv accurately, giving access to all electronic structure methods. Central to this approach is the construction of quantum(-mechanically treated) clusters placed within suitable embedding environments. Unfortunately, current approaches to constructing the quantum clusters either require large system sizes, preventing application of high-level methods, or require significant manual input, preventing investigations of multiple systems simultaneously. In this work, we present a systematic and general quantum cluster design protocol that can determine small converged quantum clusters for studying the Ov in metal-oxides with accurate methods, such as local coupled cluster with single, double, and perturbative triple excitations. We apply this protocol to study the Ov in the bulk and surface planes of rutile TiO2 and rock salt MgO, producing the first accurate and well-converged determinations of EOv with this method. These reference values are used to benchmark exchange–correlation functionals in DFT, and we find that all the studied functionals underestimate EOv, with the average error decreasing along the rungs of Jacob’s ladder. This protocol is automatable for high-throughput calculations and can be generalized to study other point defects or adsorbates.
@article{shi_general_2022, title = {General embedded cluster protocol for accurate modeling of oxygen vacancies in metal-oxides}, volume = {156}, issn = {0021-9606}, url = {https://aip.scitation.org/doi/full/10.1063/5.0087031}, doi = {10.1063/5.0087031}, number = {12}, urldate = {2022-08-19}, journal = {The Journal of Chemical Physics}, author = {Shi, Benjamin X. and Kapil, Venkat and Zen, Andrea and Chen, Ji and Alavi, Ali and Michaelides, Angelos}, month = mar, year = {2022}, pages = {124704}, csp = {true} } -
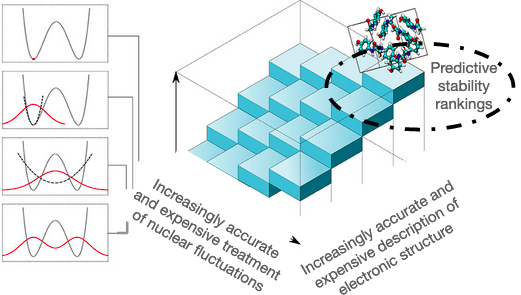 A complete description of thermodynamic stabilities of molecular crystalsVenkat Kapil, and Edgar A. EngelProceedings of the National Academy of Sciences Feb 2022
A complete description of thermodynamic stabilities of molecular crystalsVenkat Kapil, and Edgar A. EngelProceedings of the National Academy of Sciences Feb 2022Predictions of relative stabilities of (competing) molecular crystals are of great technological relevance, most notably for the pharmaceutical industry. However, they present a long-standing challenge for modeling, as often minuscule free energy differences are sensitively affected by the description of electronic structure, the statistical mechanics of the nuclei and the cell, and thermal expansion. The importance of these effects has been individually established, but rigorous free energy calculations for general molecular compounds, which simultaneously account for all effects, have hitherto not been computationally viable. Here we present an efficient “end to end” framework that seamlessly combines state-of-the art electronic structure calculations, machine-learning potentials, and advanced free energy methods to calculate ab initio Gibbs free energies for general organic molecular materials. The facile generation of machine-learning potentials for a diverse set of polymorphic compounds—benzene, glycine, and succinic acid—and predictions of thermodynamic stabilities in qualitative and quantitative agreement with experiments highlight that predictive thermodynamic studies of industrially relevant molecular materials are no longer a daunting task.
@article{kapil_complete_2022, title = {A complete description of thermodynamic stabilities of molecular crystals}, volume = {119}, copyright = {Copyright © 2022 the Author(s). Published by PNAS.. https://creativecommons.org/licenses/by/4.0/This open access article is distributed under Creative Commons Attribution License 4.0 (CC BY).}, issn = {0027-8424, 1091-6490}, url = {https://www.pnas.org/content/119/6/e2111769119}, doi = {10.1073/pnas.2111769119}, language = {en}, number = {6}, urldate = {2022-02-10}, journal = {Proceedings of the National Academy of Sciences}, author = {Kapil, Venkat and Engel, Edgar A.}, month = feb, year = {2022}, pmid = {35131847}, keywords = {Condensed Matter - Materials Science, Condensed Matter - Statistical Mechanics, ab initio thermodynamics, machine learning, polymorphism, statistical mechanics}, pi = {true}, mlps = {true}, csp = {true} }
2021
-
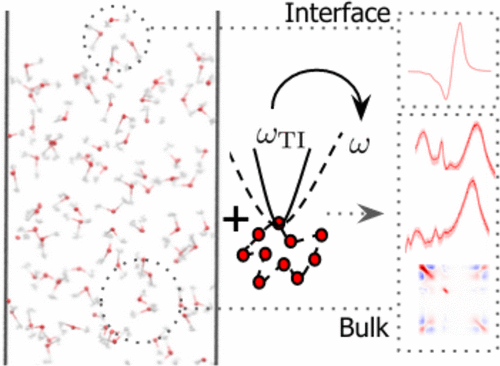 Efficient Quantum Vibrational Spectroscopy of Water with High-Order Path Integrals: From Bulk to InterfacesSam Shepherd, Jinggang Lan, David M. Wilkins, and 1 more authorThe Journal of Physical Chemistry Letters Sep 2021
Efficient Quantum Vibrational Spectroscopy of Water with High-Order Path Integrals: From Bulk to InterfacesSam Shepherd, Jinggang Lan, David M. Wilkins, and 1 more authorThe Journal of Physical Chemistry Letters Sep 2021Vibrational spectroscopy is key in probing the interplay between the structure and dynamics of aqueous systems. To map different regions of experimental spectra to the microscopic structure of a system, it is important to combine them with first-principles atomistic simulations that incorporate the quantum nature of nuclei. Here we show that the large cost of calculating the quantum vibrational spectra of aqueous systems can be dramatically reduced compared with standard path integral methods by using approximate quantum dynamics based on high-order path integrals. Together with state-of-the-art machine-learned electronic properties, our approach gives an excellent description not only of the infrared and Raman spectra of bulk water but also of the 2D correlation and the more challenging sum-frequency generation spectra of the water–air interface. This paves the way for understanding complex interfaces such as water encapsulated between or in contact with hydrophobic and hydrophilic materials through robust and inexpensive surface-sensitive and multidimensional spectra with first-principles accuracy.
@article{shepherd_efficient_2021, title = {Efficient {Quantum} {Vibrational} {Spectroscopy} of {Water} with {High}-{Order} {Path} {Integrals}: {From} {Bulk} to {Interfaces}}, shorttitle = {Efficient {Quantum} {Vibrational} {Spectroscopy} of {Water} with {High}-{Order} {Path} {Integrals}}, url = {https://doi.org/10.1021/acs.jpclett.1c02574}, doi = {10.1021/acs.jpclett.1c02574}, urldate = {2021-09-21}, journal = {The Journal of Physical Chemistry Letters}, author = {Shepherd, Sam and Lan, Jinggang and Wilkins, David M. and Kapil, Venkat}, month = sep, year = {2021}, pages = {9108--9114}, pi = {true} } -
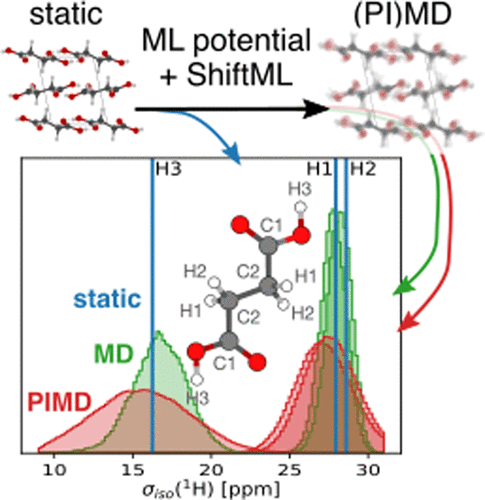 Importance of Nuclear Quantum Effects for NMR CrystallographyEdgar A. Engel, Venkat Kapil, and Michele CeriottiThe Journal of Physical Chemistry Letters Aug 2021
Importance of Nuclear Quantum Effects for NMR CrystallographyEdgar A. Engel, Venkat Kapil, and Michele CeriottiThe Journal of Physical Chemistry Letters Aug 2021The resolving power of solid-state nuclear magnetic resonance (NMR) crystallography depends heavily on the accuracy of computational predictions of NMR chemical shieldings of candidate structures, which are usually taken to be local minima in the potential energy. To test the limits of this approximation, we systematically study the importance of finite-temperature and quantum nuclear fluctuations for 1H, 13C, and 15N shieldings in polymorphs of three paradigmatic molecular crystals: benzene, glycine, and succinic acid. The effect of quantum fluctuations is comparable to the typical errors of shielding predictions for static nuclei with respect to experiments, and their inclusion improves the agreement with measurements, translating to more reliable assignment of the NMR spectra to the correct candidate structure. The use of integrated machine-learning models, trained on first-principles energies and shieldings, renders rigorous sampling of nuclear fluctuations affordable, setting a new standard for the calculations underlying NMR structure determinations.
@article{engel_importance_2021, title = {Importance of {Nuclear} {Quantum} {Effects} for {NMR} {Crystallography}}, volume = {12}, url = {https://doi.org/10.1021/acs.jpclett.1c01987}, doi = {10.1021/acs.jpclett.1c01987}, number = {32}, urldate = {2021-09-14}, journal = {The Journal of Physical Chemistry Letters}, author = {Engel, Edgar A. and Kapil, Venkat and Ceriotti, Michele}, month = aug, year = {2021}, pages = {7701--7707}, } -
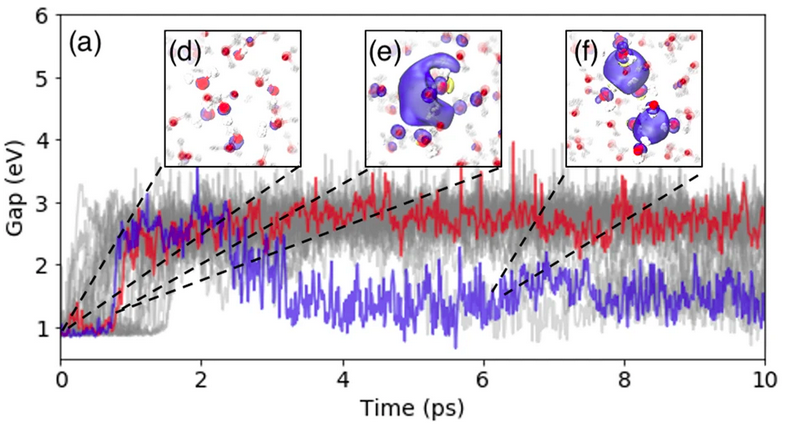 Simulating the ghost: quantum dynamics of the solvated electronJinggang Lan, Venkat Kapil, Piero Gasparotto, and 3 more authorsNature Communications Feb 2021
Simulating the ghost: quantum dynamics of the solvated electronJinggang Lan, Venkat Kapil, Piero Gasparotto, and 3 more authorsNature Communications Feb 2021The nature of the bulk hydrated electron has been a challenge for both experiment and theory due to its short lifetime and high reactivity, and the need for a high-level of electronic structure theory to achieve predictive accuracy. The lack of a classical atomistic structural formula makes it exceedingly difficult to model the solvated electron using conventional empirical force fields, which describe the system in terms of interactions between point particles associated with atomic nuclei. Here we overcome this problem using a machine-learning model, that is sufficiently flexible to describe the effect of the excess electron on the structure of the surrounding water, without including the electron in the model explicitly. The resulting potential is not only able to reproduce the stable cavity structure but also recovers the correct localization dynamics that follow the injection of an electron in neat water. The machine learning model achieves the accuracy of the state-of-the-art correlated wave function method it is trained on. It is sufficiently inexpensive to afford a full quantum statistical and dynamical description and allows us to achieve accurate determination of the structure, diffusion mechanisms, and vibrational spectroscopy of the solvated electron.
@article{lan_simulating_2021, title = {Simulating the ghost: quantum dynamics of the solvated electron}, volume = {12}, copyright = {2021 The Author(s)}, issn = {2041-1723}, shorttitle = {Simulating the ghost}, url = {https://www.nature.com/articles/s41467-021-20914-0}, doi = {10.1038/s41467-021-20914-0}, language = {en}, number = {1}, urldate = {2021-07-29}, journal = {Nature Communications}, author = {Lan, Jinggang and Kapil, Venkat and Gasparotto, Piero and Ceriotti, Michele and Iannuzzi, Marcella and Rybkin, Vladimir V.}, month = feb, year = {2021}, pages = {766}, pi = {true} } -
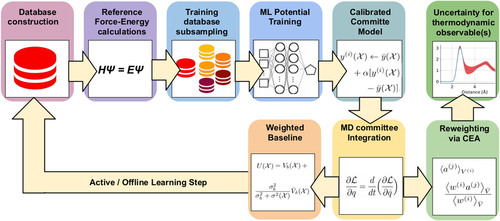 Uncertainty estimation for molecular dynamics and samplingGiulio Imbalzano, Yongbin Zhuang, Venkat Kapil, and 4 more authorsThe Journal of Chemical Physics Feb 2021
Uncertainty estimation for molecular dynamics and samplingGiulio Imbalzano, Yongbin Zhuang, Venkat Kapil, and 4 more authorsThe Journal of Chemical Physics Feb 2021Machine-learning models have emerged as a very effective strategy to sidestep time-consuming electronic-structure calculations, enabling accurate simulations of greater size, time scale, and complexity. Given the interpolative nature of these models, the reliability of predictions depends on the position in phase space, and it is crucial to obtain an estimate of the error that derives from the finite number of reference structures included during model training. When using a machine-learning potential to sample a finite-temperature ensemble, the uncertainty on individual configurations translates into an error on thermodynamic averages and leads to a loss of accuracy when the simulation enters a previously unexplored region. Here, we discuss how uncertainty quantification can be used, together with a baseline energy model, or a more robust but less accurate interatomic potential, to obtain more resilient simulations and to support active-learning strategies. Furthermore, we introduce an on-the-fly reweighing scheme that makes it possible to estimate the uncertainty in thermodynamic averages extracted from long trajectories. We present examples covering different types of structural and thermodynamic properties and systems as diverse as water and liquid gallium.
@article{imbalzano_uncertainty_2021, title = {Uncertainty estimation for molecular dynamics and sampling}, volume = {154}, issn = {0021-9606}, url = {https://aip.scitation.org/doi/10.1063/5.0036522}, doi = {10.1063/5.0036522}, number = {7}, urldate = {2021-05-31}, journal = {The Journal of Chemical Physics}, author = {Imbalzano, Giulio and Zhuang, Yongbin and Kapil, Venkat and Rossi, Kevin and Engel, Edgar A. and Grasselli, Federico and Ceriotti, Michele}, month = feb, year = {2021}, pages = {074102}, mlps = {true} }
2020
-
 Inexpensive modeling of quantum dynamics using path integral generalized Langevin equation thermostatsVenkat Kapil, David M. Wilkins, Jinggang Lan, and 1 more authorThe Journal of Chemical Physics Mar 2020
Inexpensive modeling of quantum dynamics using path integral generalized Langevin equation thermostatsVenkat Kapil, David M. Wilkins, Jinggang Lan, and 1 more authorThe Journal of Chemical Physics Mar 2020The properties of molecules and materials containing light nuclei are affected by their quantum mechanical nature. Accurate modeling of these quantum nuclear effects requires computationally demanding path integral techniques. Considerable success has been achieved in reducing the cost of such simulations by using generalized Langevin dynamics to induce frequency-dependent fluctuations. Path integral generalized Langevin equation methods, however, have this far been limited to the study of static, thermodynamic properties due to the large perturbation to the system’s dynamics induced by the aggressive thermostatting. Here, we introduce a post-processing scheme, based on analytical estimates of the dynamical perturbation induced by the generalized Langevin dynamics, which makes it possible to recover meaningful time correlation properties from a thermostatted trajectory. We show that this approach yields spectroscopic observables for model and realistic systems that have an accuracy comparable to much more demanding approximate quantum dynamics techniques based on full path integral simulations.
@article{kapil_inexpensive_2020, title = {Inexpensive modeling of quantum dynamics using path integral generalized {Langevin} equation thermostats}, volume = {152}, issn = {0021-9606}, url = {https://aip.scitation.org/doi/10.1063/1.5141950}, doi = {10.1063/1.5141950}, number = {12}, urldate = {2020-10-28}, journal = {The Journal of Chemical Physics}, author = {Kapil, Venkat and Wilkins, David M. and Lan, Jinggang and Ceriotti, Michele}, month = mar, year = {2020}, pages = {124104}, pi = {true} } -
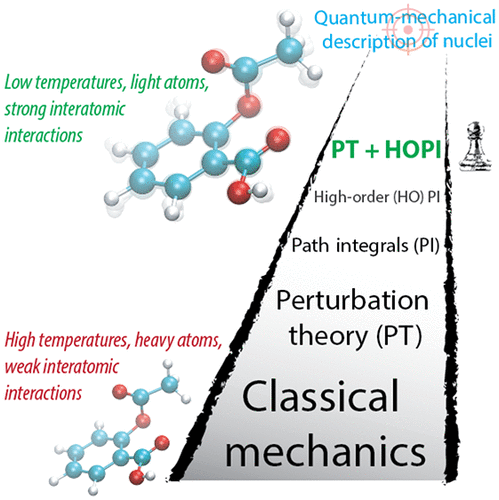 Accurate description of nuclear quantum effects with high-order perturbed path integrals (HOPPI)Igor Poltavsky, Venkat Kapil, Michele Ceriotti, and 2 more authorsJournal of Chemical Theory and Computation Jan 2020
Accurate description of nuclear quantum effects with high-order perturbed path integrals (HOPPI)Igor Poltavsky, Venkat Kapil, Michele Ceriotti, and 2 more authorsJournal of Chemical Theory and Computation Jan 2020Imaginary time path-integral (PI) simulations that account for nuclear quantum effects (NQE) beyond the harmonic approximation are increasingly employed together with modern electronic-structure calculations. Existing PI methods are applicable to molecules, liquids, and solids, however the computational cost of such simulations increases dramatically with decreasing temperature. To address this challenge, here we propose to combine high-order PI factorization with perturbation theory (PT). Already for conventional second-order PI simulations, the PT ansatz increases the accuracy twofold compared to fourth-order schemes with the same settings. In turn, applying PT to high-order path-integrals (HOPI) further improves the efficiency of simulations for molecular and condensed matter systems especially at low temperatures. We present results for bulk liquid water, aspirin molecule, and CH5+ molecule. Perturbed HOPI simulations remain both efficient and accurate down to 20 K and provide a convenient method to estimate the convergence of quantum-mechanical observables.
@article{poltavsky_accurate_2020, title = {Accurate description of nuclear quantum effects with high-order perturbed path integrals ({HOPPI})}, issn = {1549-9618}, url = {https://doi.org/10.1021/acs.jctc.9b00881}, doi = {10.1021/acs.jctc.9b00881}, urldate = {2020-01-22}, journal = {Journal of Chemical Theory and Computation}, author = {Poltavsky, Igor and Kapil, Venkat and Ceriotti, Michele and Kim, Kwang S. and Tkatchenko, Alexandre}, month = jan, year = {2020}, }
2019
-
 i-PI 2.0: A universal force engine for advanced molecular simulationsVenkat Kapil, Mariana Rossi, Ondrej Marsalek, and 24 more authorsComputer Physics Communications Mar 2019
i-PI 2.0: A universal force engine for advanced molecular simulationsVenkat Kapil, Mariana Rossi, Ondrej Marsalek, and 24 more authorsComputer Physics Communications Mar 2019Progress in the atomic-scale modeling of matter over the past decade has been tremendous. This progress has been brought about by improvements in methods for evaluating interatomic forces that work by either solving the electronic structure problem explicitly, or by computing accurate approximations of the solution and by the development of techniques that use the Born–Oppenheimer (BO) forces to move the atoms on the BO potential energy surface. As a consequence of these developments it is now possible to identify stable or metastable states, to sample configurations consistent with the appropriate thermodynamic ensemble, and to estimate the kinetics of reactions and phase transitions. All too often, however, progress is slowed down by the bottleneck associated with implementing new optimization algorithms and/or sampling techniques into the many existing electronic-structure and empirical-potential codes. To address this problem, we are thus releasing a new version of the i-PI software. This piece of software is an easily extensible framework for implementing advanced atomistic simulation techniques using interatomic potentials and forces calculated by an external driver code. While the original version of the code (Ceriotti et al., 2014) was developed with a focus on path integral molecular dynamics techniques, this second release of i-PI not only includes several new advanced path integral methods, but also offers other classes of algorithms. In other words, i-PI is moving towards becoming a universal force engine that is both modular and tightly coupled to the driver codes that evaluate the potential energy surface and its derivatives. Program summary Program Title: i-PI Program Files doi: http://dx.doi.org/10.17632/x792grbm9g.1 Licensing provisions: GPLv3, MIT Programming language: Python External routines/libraries: NumPy Nature of problem: Lowering the implementation barrier to bring state-of-the-art sampling and atomistic modeling techniques to ab initio and empirical potentials programs. Solution method: Advanced sampling methods, including path-integral molecular dynamics techniques, are implemented in a Python interface. Any electronic structure code can be patched to receive the atomic coordinates from the Python interface, and to return the forces and energy that are used to integrate the equations of motion, optimize atomic geometries, etc. Restrictions: This code does not compute interatomic potentials, although the distribution includes sample driver codes that can be used to test different techniques using a few simple model force fields.
@article{kapil_i-pi_2019, title = {i-{PI} 2.0: {A} universal force engine for advanced molecular simulations}, volume = {236}, issn = {0010-4655}, shorttitle = {i-{PI} 2.0}, url = {http://www.sciencedirect.com/science/article/pii/S0010465518303436}, doi = {10.1016/j.cpc.2018.09.020}, language = {en}, urldate = {2019-12-04}, journal = {Computer Physics Communications}, author = {Kapil, Venkat and Rossi, Mariana and Marsalek, Ondrej and Petraglia, Riccardo and Litman, Yair and Spura, Thomas and Cheng, Bingqing and Cuzzocrea, Alice and Meißner, Robert H. and Wilkins, David M. and Helfrecht, Benjamin A. and Juda, Przemysław and Bienvenue, Sébastien P. and Fang, Wei and Kessler, Jan and Poltavsky, Igor and Vandenbrande, Steven and Wieme, Jelle and Corminboeuf, Clemence and Kühne, Thomas D. and Manolopoulos, David E. and Markland, Thomas E. and Richardson, Jeremy O. and Tkatchenko, Alexandre and Tribello, Gareth A. and Van Speybroeck, Veronique and Ceriotti, Michele}, month = mar, year = {2019}, keywords = {Accelerated sampling, Geometry optimizers, Molecular dynamics, Path integral}, pages = {214--223}, pi = {true} } -
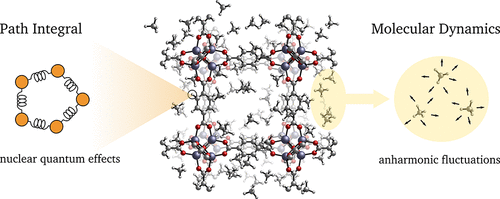 Modeling the Structural and Thermal Properties of Loaded Metal–Organic Frameworks. An Interplay of Quantum and Anharmonic FluctuationsVenkat Kapil, Jelle Wieme, Steven Vandenbrande, and 3 more authorsJournal of Chemical Theory and Computation May 2019
Modeling the Structural and Thermal Properties of Loaded Metal–Organic Frameworks. An Interplay of Quantum and Anharmonic FluctuationsVenkat Kapil, Jelle Wieme, Steven Vandenbrande, and 3 more authorsJournal of Chemical Theory and Computation May 2019Metal–organic frameworks show both fundamental interest and great promise for applications in adsorption-based technologies, such as the separation and storage of gases. The flexibility and complexity of the molecular scaffold pose a considerable challenge to atomistic modeling, especially when also considering the presence of guest molecules. We investigate the role played by quantum and anharmonic fluctuations in the archetypical case of MOF-5, comparing the material at various levels of methane loading. Accurate path integral simulations of such effects are made affordable by the introduction of an accelerated simulation scheme and the use of an optimized force field based on first-principles reference calculations. We find that the level of statistical treatment that is required for predictive modeling depends significantly on the property of interest. The thermal properties of the lattice are generally well described by a quantum harmonic treatment, with the adsorbate behaving in a classical but strongly anharmonic manner. The heat capacity of the loaded framework–which plays an important role in the characterization of the framework and in determining its stability to thermal fluctuations during adsorption/desorption cycles–requires, however, a full quantum and anharmonic treatment, either by path integral methods or by a simple but approximate scheme. We also present molecular-level insight into the nanoscopic interactions contributing to the material’s properties and suggest design principles to optimize them.
@article{kapil_modeling_2019, title = {Modeling the {Structural} and {Thermal} {Properties} of {Loaded} {Metal}–{Organic} {Frameworks}. {An} {Interplay} of {Quantum} and {Anharmonic} {Fluctuations}}, volume = {15}, issn = {1549-9618}, url = {https://doi.org/10.1021/acs.jctc.8b01297}, doi = {10.1021/acs.jctc.8b01297}, number = {5}, urldate = {2020-01-22}, journal = {Journal of Chemical Theory and Computation}, author = {Kapil, Venkat and Wieme, Jelle and Vandenbrande, Steven and Lamaire, Aran and Van Speybroeck, Veronique and Ceriotti, Michele}, month = may, year = {2019}, pages = {3237--3249}, } -
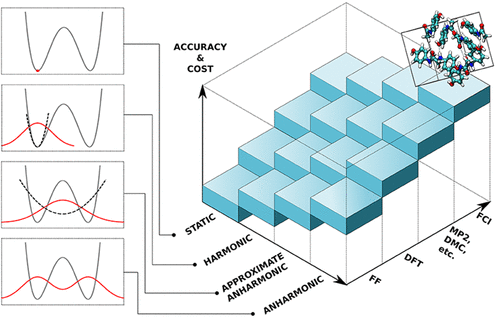 Assessment of Approximate Methods for Anharmonic Free EnergiesVenkat Kapil, Edgar Engel, Mariana Rossi, and 1 more authorJournal of Chemical Theory and Computation Nov 2019
Assessment of Approximate Methods for Anharmonic Free EnergiesVenkat Kapil, Edgar Engel, Mariana Rossi, and 1 more authorJournal of Chemical Theory and Computation Nov 2019Quantitative evaluation of the thermodynamic properties of materials—most notably their stability, as measured by the free energy—must take into account the role of thermal and zero-point energy fluctuations. While these effects can easily be estimated within a harmonic approximation, corrections arising from the anharmonic nature of the interatomic potential are often crucial and require computationally costly path integral simulations to obtain results that are essentially exact for a given potential. Consequently, different approximate frameworks for computing affordable estimates of the anharmonic free energies have been developed over the years. Understanding which of the approximations involved are justified for a given system, and therefore choosing the most suitable method, is complicated by the lack of comparative benchmarks. To facilitate this choice we assess the accuracy and efficiency of some of the most commonly used approximate methods: the independent mode framework, the vibrational self-consistent field, and self-consistent phonons. We compare the anharmonic correction to the Helmholtz free energy against reference path integral calculations. These benchmarks are performed for a diverse set of systems, ranging from simple weakly anharmonic solids to flexible molecular crystals with freely rotating units. The results suggest that, for simple solids such as allotropes of carbon, these methods yield results that are in excellent agreement with the reference calculations, at a considerably lower computational cost. For more complex molecular systems such as polymorphs of ice and paracetamol the methods do not consistently provide a reliable approximation of the anharmonic correction. Despite substantial cancellation of errors when comparing the stability of different phases, we do not observe a systematic improvement over the harmonic approximation even for relative free energies. We conclude that, at least for the classes of materials considered here, efforts toward obtaining computationally feasible anharmonic free energies should therefore be directed toward reducing the expense of path integral methods.
@article{kapil_assessment_2019, title = {Assessment of {Approximate} {Methods} for {Anharmonic} {Free} {Energies}}, volume = {15}, issn = {1549-9618}, url = {https://doi.org/10.1021/acs.jctc.9b00596}, doi = {10.1021/acs.jctc.9b00596}, number = {11}, urldate = {2020-01-22}, journal = {Journal of Chemical Theory and Computation}, author = {Kapil, Venkat and Engel, Edgar and Rossi, Mariana and Ceriotti, Michele}, month = nov, year = {2019}, pages = {5845--5857}, csp = {true} } -
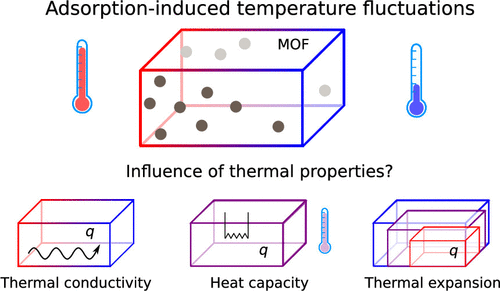 Thermal Engineering of Metal–Organic Frameworks for Adsorption Applications: A Molecular Simulation PerspectiveJelle Wieme, Steven Vandenbrande, Aran Lamaire, and 3 more authorsACS Applied Materials & Interfaces Oct 2019
Thermal Engineering of Metal–Organic Frameworks for Adsorption Applications: A Molecular Simulation PerspectiveJelle Wieme, Steven Vandenbrande, Aran Lamaire, and 3 more authorsACS Applied Materials & Interfaces Oct 2019Thermal engineering of metal–organic frameworks for adsorption-based applications is very topical in view of their industrial potential, in particular, since heat management and thermal stability have been identified as important obstacles. Hence, a fundamental understanding of the structural and chemical features underpinning their intrinsic thermal properties is highly sought-after. Herein, we investigate the nanoscale behavior of a diverse set of frameworks using molecular simulation techniques and critically compare properties such as thermal conductivity, heat capacity, and thermal expansion with other classes of materials. Furthermore, we propose a hypothetical thermodynamic cycle to estimate the temperature rise associated with adsorption for the most important greenhouse and energy-related gases (CO2 and CH4). This macroscopic response on the heat of adsorption connects the intrinsic thermal properties with the adsorption properties and allows us to evaluate their importance.
@article{wieme_thermal_2019, title = {Thermal {Engineering} of {Metal}–{Organic} {Frameworks} for {Adsorption} {Applications}: {A} {Molecular} {Simulation} {Perspective}}, volume = {11}, issn = {1944-8244}, shorttitle = {Thermal {Engineering} of {Metal}–{Organic} {Frameworks} for {Adsorption} {Applications}}, url = {https://doi.org/10.1021/acsami.9b12533}, doi = {10.1021/acsami.9b12533}, number = {42}, urldate = {2020-01-22}, journal = {ACS Applied Materials \& Interfaces}, author = {Wieme, Jelle and Vandenbrande, Steven and Lamaire, Aran and Kapil, Venkat and Vanduyfhuys, Louis and Van Speybroeck, Veronique}, month = oct, year = {2019}, pages = {38697--38707}, } -
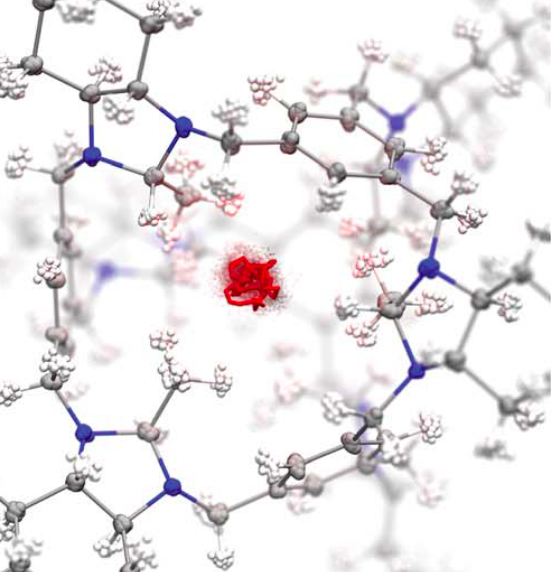 Barely porous organic cages for hydrogen isotope separationMing Liu, Linda Zhang, Marc A. Little, and 13 more authorsScience Nov 2019
Barely porous organic cages for hydrogen isotope separationMing Liu, Linda Zhang, Marc A. Little, and 13 more authorsScience Nov 2019Quantum sieves for hydrogen isotopes One method for improving the efficiency of separation of hydrogen from deuterium (D) is to exploit kinetic quantum sieving with nanoporous solids. This method requires ultrafine pore apertures (around 3 angstroms), which usually leads to low pore volumes and low D2 adsorption capacities. Liu et al. used organic synthesis to tune the pore size of the internal cavities of organic cage molecules. A hybrid cocrystal contained both a small-pore cage that imparted high selectivity and a larger-pore cage that enabled high D2 uptake. Science, this issue p. 613 The separation of hydrogen isotopes for applications such as nuclear fusion is a major challenge. Current technologies are energy intensive and inefficient. Nanoporous materials have the potential to separate hydrogen isotopes by kinetic quantum sieving, but high separation selectivity tends to correlate with low adsorption capacity, which can prohibit process scale-up. In this study, we use organic synthesis to modify the internal cavities of cage molecules to produce hybrid materials that are excellent quantum sieves. By combining small-pore and large-pore cages together in a single solid, we produce a material with optimal separation performance that combines an excellent deuterium/hydrogen selectivity (8.0) with a high deuterium uptake (4.7 millimoles per gram). Cocrystals of modified molecular organic crystals can separate hydrogen from deuterium through kinetic quantum sieving. Cocrystals of modified molecular organic crystals can separate hydrogen from deuterium through kinetic quantum sieving.
@article{liu_barely_2019, title = {Barely porous organic cages for hydrogen isotope separation}, volume = {366}, copyright = {Copyright © 2019 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works. http://www.sciencemag.org/about/science-licenses-journal-article-reuseThis is an article distributed under the terms of the Science Journals Default License.}, issn = {0036-8075, 1095-9203}, url = {https://science.sciencemag.org/content/366/6465/613}, doi = {10.1126/science.aax7427}, language = {en}, number = {6465}, urldate = {2019-11-18}, journal = {Science}, author = {Liu, Ming and Zhang, Linda and Little, Marc A. and Kapil, Venkat and Ceriotti, Michele and Yang, Siyuan and Ding, Lifeng and Holden, Daniel L. and Balderas-Xicohténcatl, Rafael and He, Donglin and Clowes, Rob and Chong, Samantha Y. and Schütz, Gisela and Chen, Linjiang and Hirscher, Michael and Cooper, Andrew I.}, month = nov, year = {2019}, pmid = {31672893}, pages = {613--620}, pi = {true} }
2018
-
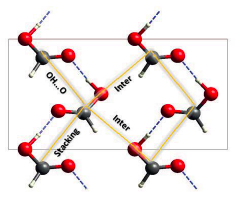 Hydrogen dynamics in solid formic acid: insights from simulations with quantum colored-noise thermostatsK. Dru}.zbicki, M. Krzystyniak, D. Hollas, and 4 more authorsJournal of Physics: Conference Series Jul 2018
Hydrogen dynamics in solid formic acid: insights from simulations with quantum colored-noise thermostatsK. Dru}.zbicki, M. Krzystyniak, D. Hollas, and 4 more authorsJournal of Physics: Conference Series Jul 2018With an increase of computational capabilities, ab initio molecular dynamics becomes the natural choice for exploring the nuclear dynamics of solids. As based on classical mechanics, the validity of this approach is, in-principle, limited to the high-T regime, whilst low-temperature simulations require inclusion of quantum effects. The methods commonly used to account for nuclear quantum effects are based on the path-integral formalism, which become, however, particularly time consuming when high accuracy methods are used for calculating forces. Recently, new efficient alternative approaches to account for quantum nature of nuclei have been proposed, using so-called quantum thermostats. In this work, we examine the simulations performed with the quantum colored-noise thermostat introduced by Ceriotti [Phys. Rev. Lett., 103:030603, 2009]. We present the tests of portable implementation of the quantum thermostat in the ABIN program, which has been extended to periodic systems through the interface to CASTEP, a leading spectroscopy-oriented plane-wave density functional theory code. The range of applicability of quantum-thermostatted molecular dynamics simulations for the interpretation of neutron scattering data was examined and compared to classical molecular dynamics and lattice-dynamics simulations, using solid formic acid case as a test bed. We find that the approach is particularly useful for the modeling of low-temperature inelastic neutron scattering spectra as well as provides some theoretical estimate for the low-limit of the mean kinetic energy. While finding the quantum-thermostat to seriously affect the dynamic properties of the title system, we illustrate to which extent the unperturbed response can be successfully recovered.
@article{druzbicki_hydrogen_2018, title = {Hydrogen dynamics in solid formic acid: insights from simulations with quantum colored-noise thermostats}, volume = {1055}, issn = {1742-6596}, shorttitle = {Hydrogen dynamics in solid formic acid}, url = {https://doi.org/10.1088/1742-6596/1055/1/012003}, doi = {10.1088/1742-6596/1055/1/012003}, language = {en}, urldate = {2022-08-19}, journal = {Journal of Physics: Conference Series}, author = {Dru{\textbackslash}.zbicki, K. and Krzystyniak, M. and Hollas, D. and Kapil, V. and Slavíček, P. and Romanelli, G. and Fernandez-Alonso, F.}, month = jul, year = {2018}, pages = {012003}, } -
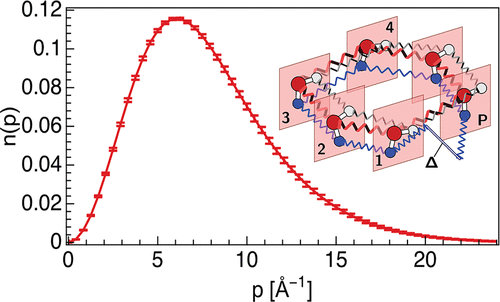 Anisotropy of the Proton Momentum Distribution in WaterVenkat Kapil, Alice Cuzzocrea, and Michele CeriottiThe Journal of Physical Chemistry B Jun 2018
Anisotropy of the Proton Momentum Distribution in WaterVenkat Kapil, Alice Cuzzocrea, and Michele CeriottiThe Journal of Physical Chemistry B Jun 2018One of the many peculiar properties of water is the pronounced deviation of the proton momentum distribution from Maxwell–Boltzmann behavior. This deviation from the classical limit is a manifestation of the quantum mechanical nature of protons. Its extent, which can be probed directly by deep inelastic neutron scattering experiments, gives important insight on the potential of mean force felt by H atoms. The determination of the full distribution of particle momenta, however, is a real tour de force for both experiments and theory, which has led to unresolved discrepancies between the two. In this study, we present comprehensive, fully converged momentum distributions for water at several thermodynamic state points, focusing on the components that cannot be described in terms of a scalar contribution to the quantum kinetic energy, and providing a benchmark that can serve as a reference for future simulations and experiments. In doing so, we also introduce a number of technical developments that simplify and accelerate greatly the calculation of momentum distributions by means of atomistic simulations.
@article{kapil_anisotropy_2018, title = {Anisotropy of the {Proton} {Momentum} {Distribution} in {Water}}, volume = {122}, issn = {1520-6106}, url = {https://doi.org/10.1021/acs.jpcb.8b03896}, doi = {10.1021/acs.jpcb.8b03896}, number = {22}, urldate = {2020-01-22}, journal = {The Journal of Physical Chemistry B}, author = {Kapil, Venkat and Cuzzocrea, Alice and Ceriotti, Michele}, month = jun, year = {2018}, pages = {6048--6054}, }
2017
-
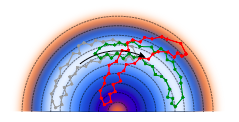 Fine tuning classical and quantum molecular dynamics using a generalized Langevin equationMariana Rossi, Venkat Kapil, and Michele CeriottiThe Journal of Chemical Physics Jul 2017
Fine tuning classical and quantum molecular dynamics using a generalized Langevin equationMariana Rossi, Venkat Kapil, and Michele CeriottiThe Journal of Chemical Physics Jul 2017Generalized Langevin Equation (GLE) thermostats have been used very effectively as a tool to manipulate and optimize the sampling of thermodynamic ensembles and the associated static properties. Here we show that a similar, exquisite level of control can be achieved for the dynamical properties computed from thermostatted trajectories. We develop quantitative measures of the disturbance induced by the GLE to the Hamiltonian dynamics of a harmonic oscillator, and show that these analytical results accurately predict the behavior of strongly anharmonic systems. We also show that it is possible to correct, to a significant extent, the effects of the GLE term onto the corresponding microcanonical dynamics, which puts on more solid grounds the use of non-equilibrium Langevin dynamics to approximate quantum nuclear effects and could help improve the prediction of dynamical quantities from techniques that use a Langevin term to stabilize dynamics. Finally we address the use of thermostats in the context of approximate path-integral-based models of quantum nuclear dynamics. We demonstrate that a custom-tailored GLE can alleviate some of the artifacts associated with these techniques, improving the quality of results for the modeling of vibrational dynamics of molecules, liquids, and solids.
@article{rossi_fine_2017, title = {Fine tuning classical and quantum molecular dynamics using a generalized {Langevin} equation}, volume = {148}, issn = {0021-9606}, url = {https://aip.scitation.org/doi/10.1063/1.4990536}, doi = {10.1063/1.4990536}, number = {10}, urldate = {2019-10-29}, journal = {The Journal of Chemical Physics}, author = {Rossi, Mariana and Kapil, Venkat and Ceriotti, Michele}, month = jul, year = {2017}, pages = {102301}, }
2016
-
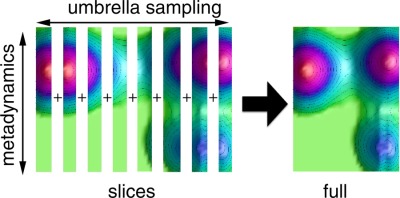 Sampling free energy surfaces as slices by combining umbrella sampling and metadynamicsShalini Awasthi, Venkat Kapil, and Nisanth N. NairJournal of Computational Chemistry Jul 2016
Sampling free energy surfaces as slices by combining umbrella sampling and metadynamicsShalini Awasthi, Venkat Kapil, and Nisanth N. NairJournal of Computational Chemistry Jul 2016Metadynamics (MTD) is a very powerful technique to sample high-dimensional free energy landscapes, and due to its self-guiding property, the method has been successful in studying complex reactions and conformational changes. MTD sampling is based on filling the free energy basins by biasing potentials and thus for cases with flat, broad, and unbound free energy wells, the computational time to sample them becomes very large. To alleviate this problem, we combine the standard Umbrella Sampling (US) technique with MTD to sample orthogonal collective variables (CVs) in a simultaneous way. Within this scheme, we construct the equilibrium distribution of CVs from biased distributions obtained from independent MTD simulations with umbrella potentials. Reweighting is carried out by a procedure that combines US reweighting and Tiwary–Parrinello MTD reweighting within the Weighted Histogram Analysis Method (WHAM). The approach is ideal for a controlled sampling of a CV in a MTD simulation, making it computationally efficient in sampling flat, broad, and unbound free energy surfaces. This technique also allows for a distributed sampling of a high-dimensional free energy surface, further increasing the computational efficiency in sampling. We demonstrate the application of this technique in sampling high-dimensional surface for various chemical reactions using ab initio and QM/MM hybrid molecular dynamics simulations. Further, to carry out MTD bias reweighting for computing forward reaction barriers in ab initio or QM/MM simulations, we propose a computationally affordable approach that does not require recrossing trajectories. © 2016 Wiley Periodicals, Inc.
@article{awasthi_sampling_2016, title = {Sampling free energy surfaces as slices by combining umbrella sampling and metadynamics}, volume = {37}, issn = {1096-987X}, url = {https://onlinelibrary.wiley.com/doi/abs/10.1002/jcc.24349}, doi = {10.1002/jcc.24349}, language = {en}, number = {16}, urldate = {2022-08-19}, journal = {Journal of Computational Chemistry}, author = {Awasthi, Shalini and Kapil, Venkat and Nair, Nisanth N.}, year = {2016}, keywords = {free energy calculations, metadynamics, reweighting, umbrella sampling, weighted histogram analysis}, pages = {1413--1424}, } -
 Accurate molecular dynamics and nuclear quantum effects at low cost by multiple steps in real and imaginary time: Using density functional theory to accelerate wavefunction methodsV. Kapil, J. VandeVondele, and M. CeriottiThe Journal of Chemical Physics Feb 2016
Accurate molecular dynamics and nuclear quantum effects at low cost by multiple steps in real and imaginary time: Using density functional theory to accelerate wavefunction methodsV. Kapil, J. VandeVondele, and M. CeriottiThe Journal of Chemical Physics Feb 2016The development and implementation of increasingly accurate methods for electronic structure calculations mean that, for many atomistic simulation problems, treating light nuclei as classical particles is now one of the most serious approximations. Even though recent developments have significantly reduced the overhead for modeling the quantum nature of the nuclei, the cost is still prohibitive when combined with advanced electronic structure methods. Here we present how multiple time step integrators can be combined with ring-polymer contraction techniques (effectively, multiple time stepping in imaginary time) to reduce virtually to zero the overhead of modelling nuclear quantum effects, while describing inter-atomic forces at high levels of electronic structure theory. This is demonstrated for a combination of MP2 and semi-local DFT applied to the Zundel cation. The approach can be seamlessly combined with other methods to reduce the computational cost of path integral calculations, such as high-order factorizations of the Boltzmann operator or generalized Langevin equation thermostats.
@article{kapil_accurate_2016, title = {Accurate molecular dynamics and nuclear quantum effects at low cost by multiple steps in real and imaginary time: {Using} density functional theory to accelerate wavefunction methods}, volume = {144}, issn = {0021-9606}, shorttitle = {Accurate molecular dynamics and nuclear quantum effects at low cost by multiple steps in real and imaginary time}, url = {https://aip.scitation.org/doi/10.1063/1.4941091}, doi = {10.1063/1.4941091}, number = {5}, urldate = {2020-01-22}, journal = {The Journal of Chemical Physics}, author = {Kapil, V. and VandeVondele, J. and Ceriotti, M.}, month = feb, year = {2016}, pages = {054111}, } -
 High order path integrals made easyVenkat Kapil, Jörg Behler, and Michele CeriottiThe Journal of Chemical Physics Dec 2016
High order path integrals made easyVenkat Kapil, Jörg Behler, and Michele CeriottiThe Journal of Chemical Physics Dec 2016The precise description of quantum nuclear fluctuations in atomistic modelling is possible by employing path integral techniques, which involve a considerable computational overhead due to the need of simulating multiple replicas of the system. Many approaches have been suggested to reduce the required number of replicas. Among these, high-order factorizations of the Boltzmann operator are particularly attractive for high-precision and low-temperature scenarios. Unfortunately, to date, several technical challenges have prevented a widespread use of these approaches to study the nuclear quantum effects in condensed-phase systems. Here we introduce an inexpensive molecular dynamics scheme that overcomes these limitations, thus making it possible to exploit the improved convergence of high-order path integrals without having to sacrifice the stability, convenience, and flexibility of conventional second-order techniques. The capabilities of the method are demonstrated by simulations of liquid water and ice, as described by a neural-network potential fitted to the dispersion-corrected hybrid density functional theory calculations.
@article{kapil_high_2016, title = {High order path integrals made easy}, volume = {145}, issn = {0021-9606}, url = {https://aip.scitation.org/doi/10.1063/1.4971438}, doi = {10.1063/1.4971438}, number = {23}, urldate = {2019-11-19}, journal = {The Journal of Chemical Physics}, author = {Kapil, Venkat and Behler, Jörg and Ceriotti, Michele}, month = dec, year = {2016}, pages = {234103}, }